
Drug Development Process
The drug development process includes five core steps:
- Discovery and development
- Preclinical research
- Clinical research
- FDA review
- FDA post-market safety monitoring
Discovery and Development
At this stage, thousands of compounds may be potential candidates for the development of medical treatments. However, only a small number of compounds will look promising enough to pursue further study and evaluation. Once researchers identify a potentially promising new compound, they typically conduct experiments to gather information pertaining to: how it is absorbed, distributed, metabolized, and excreted; its potential benefits and mechanisms of action; the optimal dosage; the best way to administer the drug; side effects or adverse events (toxicity); how it may affect different groups of people (such as people of different genders, races, or ethnicities); how it interacts with other drugs and medical treatments; and its effectiveness, as compared with similar drugs.[1]
Preclinical Research
Before a drug candidate can be tested in humans, researchers must determine toxicity—or whether it has the potential to cause serious harm. There are two types of preclinical research: 1) in vitro and 2) in vivo. Preclinical studies are typically on the smaller side. However, these studies must provide detailed information pertaining to dosing and toxicity levels. After preclinical testing, researchers review their findings to determine whether the drug should be tested in humans.[2] The FDA requires researchers to use good laboratory practices, the regulations for which are found in 21 CFR Part 58.1: Good Laboratory Practice for Nonclinical Laboratory Studies.
Clinical Research
The clinical research phase pertains to the studies (clinical trials) performed with humans. Developers first design the clinical study, taking into consideration exactly what they want to accomplish for each of the different Clinical Research Phases and beginning with the Investigational New Drug Process (IND)—a process that all drug developers must go through before a clinical trial can begin. In designing a clinical trial, researchers review information about the drug to then develop research questions and objectives. They decide:
- Who qualifies for the clinical trial (selection criteria)
- How many people will be part of the study
- How long the study will last
- Whether there will be a control group, or other ways to limit research bias
- How the drug will be administered to patients, and at what dosage
- What assessments will be conducted, when, and the data that will be collected
- How the data will be reviewed and analyzed
Clinical trials can fall into one of four phases and they typically follow a progression from early, small-scale Phase 1 studies to late-stage and large-scale Phase 3 studies. The purpose of a Phase 1 clinical trial is to assess safety and dosage. Phase 1 clinical trials recruit approximately 20-100 human subjects (healthy volunteers or people with the disease/condition) and the trial can last several months. About 70% of drugs that undergo a Phase 1 clinical trial progress to Phase 2. Phase 2 clinical trials focus on efficacy and side effects. A Phase 2 clinical trial recruits up to several hundred participants with the disease or condition and the duration of the clinical trial can last from several months to two years. Approximately 33% of drugs in Phase 2 clinical trials move on to the next phase. Phase 3 clinical trials focus on efficacy and monitoring for adverse reactions. A typical Phase 3 clinical trial will recruit approximately 300-3,000 volunteers who have the disease or condition and the length of the study lasts from 1-4 years. Approximately 25-30% of the drugs in Phase 3 clinical trials will move on to Phase 4. A Phase 4 clinical trial focuses on safety and efficacy. These trials recruit several thousand volunteers who have the disease or condition.
Before beginning any clinical research, drug developers much submit an Investigational New Drug (IND) application to the FDA. In the IND application, developers must include:
- Animal study data and toxicity (side effects that can be harmful) data
- Manufacturing information
- Clinical protocols (study plans) for studies that are to be conducted
- Data from any prior human research
- Information about the investigator
An FDA IND Review Team—comprised of a project manager, medical officer, statistician, pharmacologist, pharmakineticist, chemist, and microbiologist—has 30 days to review the original IND submission. The FDA IND Review team will either approve the clinical trial to begin or they will place a clinical hold to delay or stop the investigation. The developer is responsible for keeping the FDA team up-to-date regarding any new protocols or serious side effects observed during the trial. After the trial ends, researchers must submit study reports. The process continues until either the developer decides to end clinical trials or the developer files a marketing application. In order to file a marketing application, drug developers must have “adequate data from two large, controlled clinical trials.”[3]
FDA Drug Review
If a drug developer has evidence from early tests, preclinical research, and clinical research that a drug is safe and effective for its intended use, the company can then file an application to market the drug. A New Drug Application (NDA) includes all information about the drug—from preclinical data to Phase 3 trial data. Developers must include reports on all studies, data, and analysis. Along with clinical results, drug developers must also include proposed labeling, safety updates, drug abuse information, patent information, any data from studies conducted outside of the US, institutional review board compliance information, and directions for use. Once the FDA receives an NDA, the first step is to determine if the NDA is complete. If it is complete, the review team has approximately 6-10 months to make a decision on whether or not to approve the drug.[4] For more on how the FDA is Speeding Up the Approval Process. Moreover the FDA has developed four distinct and successful approaches to making such drugs available as rapidly as possible, including:
- Priority Review
- Breakthrough Therapy
- Accelerated Approval
- Fast Track [5]

Source: FDA
FDA Post-Market Drug Safety Monitoring
While clinical trials provide insight into a drug’s efficacy and safety, the FDA continues to monitor for drug safety over the course of the lifetime that the product is on the market. Despite the very rigorous drug approval process, it is recognized that there are limitations to this approach and that a true picture of a drug’s safety may evolve over a period of months or years. The FDA reviews reports of issues with both prescription and OTC drugs and the agency can decide to add cautions to the dosage or usage information.[6]
Office of New Drugs
The Office of New Drugs is an office within the FDA’s Center for Drug Evaluation and Research. The core mission of the FDA’s Office of New Drugs (OND) is to “ensure that safe and effective drugs and biologics are available to the American people.”[7] The OND is responsible for reviewing applications and making drug approval decisions, as well as determining and following guidance and policy to support an efficient review process. This office provides regulatory oversight for investigational studies during the drug development phase, makes decisions regarding market approval for new drugs (including changes to drugs that are already on the market), and regulates over-the-counter drugs.[7]
General contact information for the FDA’s Office of New Drugs:
Office of New Drugs
Immediate Office – Mail Stop 6311
10903 New Hampshire Avenue
Silver Spring, MD 20993
(301) 796-0700
ONDCommunications@fda.hhs.gov
The OND Office and Division contact information is available here. Specific points of contact are also listed below.
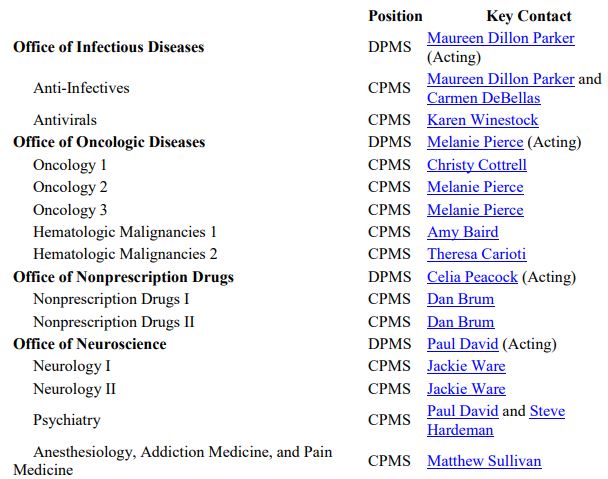
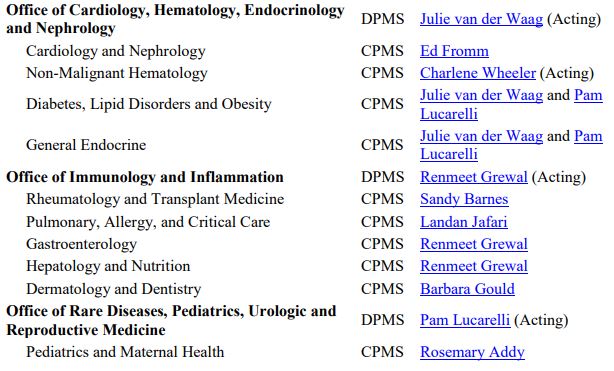

Source: FDA OND
Updated by Theresa Pipher, November 2020
Comments are closed.